Blog
Bayer hosts ChemIntelligence at the LifeHub Lyon

Joint press release from ChemIntelligence and Bayer.
DeepMatter acquires ChemIntelligence

We are pleased to announce that ChemIntelligence got acquired by DeepMatter Group Plc.
Machine learning descriptors for molecules
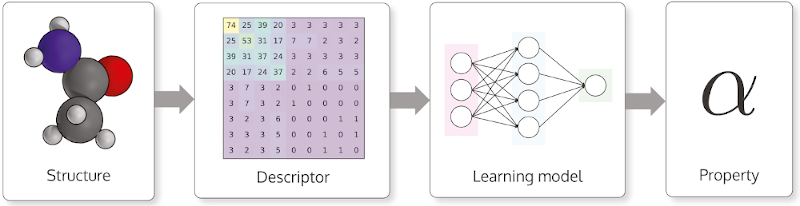
Using machine learning to predict properties of molecules or to design molecules with desired properties plays an important role in the field of chemistry, toxicology and materials science. In machine learning, when it comes to predicting the properties of molecules, it is necessary to convert the molecular structures into values that can be used by machine learning algorithms.
Developing self-optimizing reactors : The SMAPI project
![]()
We are delighted to announce that the collaborative project "SMAPI", which involves ChemIntelligence and three other partners, has been accepted for funding by the Région Auvergne-Rhône-Alpes.
Active learning for fast discovery of materials with targeted properties
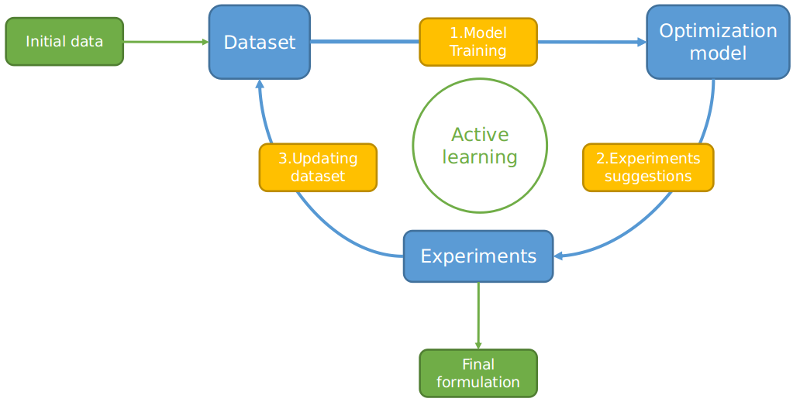
Designing new materials with targeted properties is often a long process, as there can be huge numbers of possible compositions and researchers often solely rely on their experience and on trial-and-error testing to find the right ones. Xue et al. wrote an article titled "Accelerated search for materials with targeted properties by adaptive design", in which they described how they tried to obtain NiTi-based shape-memory alloys with very low thermal hysteresis. They calculated that there were ~800 000 potential alloys in their search space. With no efficient physical models and simulation tools available to them, finding the desired alloy could have been a struggle. Fortunately, a solution was rapidly found thanks to active learning.
The different sources of data that can be used for machine learning applied to chemistry and materials R&D
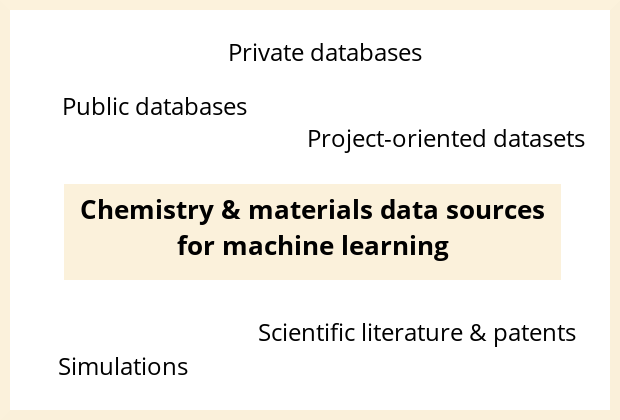
Machine learning is a powerful tool to accelerate chemistry and material science R&D as it allows to find hidden trends in data, making it possible to predict the outcome of experiments or to suggest experiments to achieve an objective (for example, maximizing the yield of a synthesis). When considering using machine learning for chemistry or material science R&D, one of the first questions to be asked is: What data can be used and should be used?
Machine-learning-assisted materials discovery using failed experiments
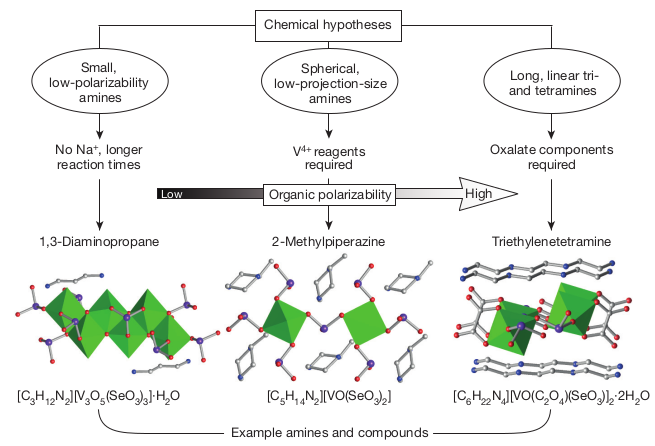
Failed experiments are not useless. They even have a lot of value! Raccuglia et al. have published an article titled "Machine-learning-assisted materials discovery using failed experiments" (Nature 2016, 533, 73-76), in which they present how they exploited failed or unsuccessful attempts at synthesizing vanadium selenites to train a machine learning program to predict the outcomes of the syntheses of other vanadium selenites with never-tested organic building blocks. The authors also studied how the machine learning model made its predictions and revealed new hypotheses about the requirements for successful synthesis of templated vanadium selenites. Their methodology is summarized in the figure below.
Using Machine Learning To Predict Suitable Conditions for Organic Reactions
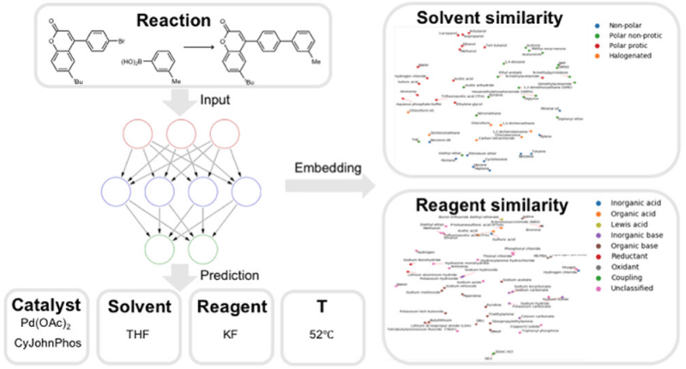
Finding the right experimental conditions (solvent, temperature, catalyst, additives, etc.) for a reaction can be a very time-consuming process. Gao et al. have published an article titled "Using Machine Learning To Predict Suitable Conditions for Organic Reactions" (ACS Cent. Sci. 2018, 4, 1465-1476), in which they present a neural network model that is able to predict appropriate reaction conditions for any organic reaction, including a catalyst, solvents, reagents and the temperature.
A graph-convolutional neural network model for the prediction of chemical reactivity
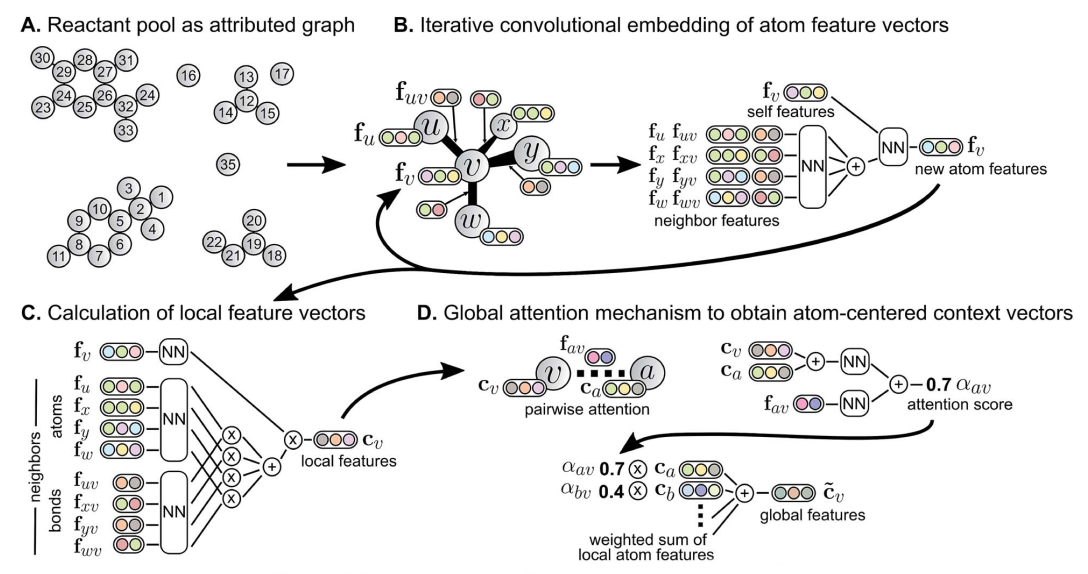
To synthesize a molecule, a chemist has to imagine a sequence of possible chemical transformations that could produce it, based on his/her knowledge and the scientific literature, and then perform the reactions in a laboratory, hoping that they happen as expected and give the desired product. Any chemist who has spent some time in a laboratory attempting to synthesize molecules knows that chemical reactions often behave in unwanted ways:
Deep-learning-based inverse design model for intelligent discovery of organic molecules
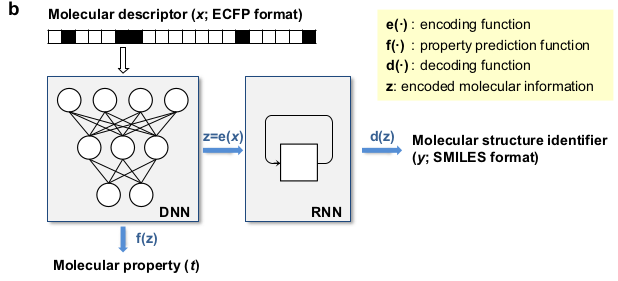
Discovering new organic molecules that possess a given property is not an easy task. It is a process that is iterative, experiment-intensive and tedious. Furthermore, designing large numbers (let's say hundreds or thousands of them) of new molecules for an application is extremely challenging, even for the most creative chemists. In our last blog post, we presented an AI-based tool that is able to automatically design new molecules, based on a continuous representation of molecules.
Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules
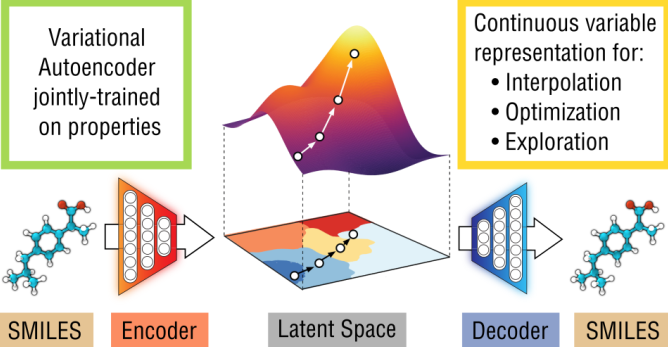
Designing molecules that have desired properties is a long and difficult process. To obtain a new molecule that can be used in some application, scientists must use their creativity and domain knowledge to propose many new molecules, synthesize them and test them for the given application. Moreover, human creativity often has its limits in the number and diversity of ideas that it can generate.
Accelerated discovery of metallic glasses through iteration of machine learning and high-throughput experiments
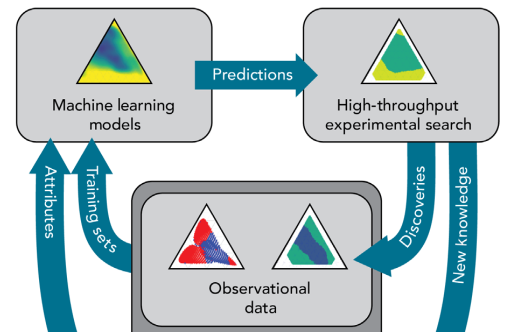
Metallic glasses are amorphous alloys of metals and metalloids. They usually have properties that are very different from crystalline alloys: exceptional mechanical performances (eg. yield strength and wear resistance) and sometimes improved corrosion resistance or high magnetic permeability.
Optimizing Chemical Reactions with Deep Reinforcement Learning
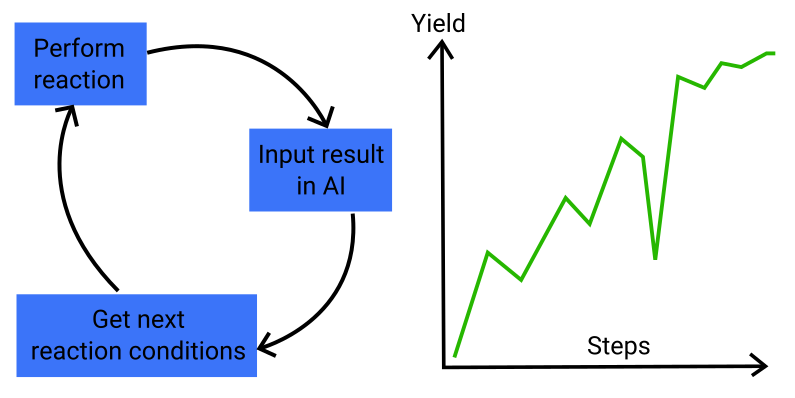
Optimizing chemical reactions is a very common task for chemists. It usually aims at maximizing the yield or selectivity of a reaction in order to get the most possible product from some raw material.
Launching ChemIntelligence
![]()
Artificial intelligence (AI) will change the way we make chemistry and materials and make it faster, by better targeting experiments that we run in the laboratory. This will allow to increase the return on investment of chemistry and materials R&D.