A graph-convolutional neural network model for the prediction of chemical reactivity
To synthesize a molecule, a chemist has to imagine a sequence of possible chemical transformations that could produce it, based on his/her knowledge and the scientific literature, and then perform the reactions in a laboratory, hoping that they happen as expected and give the desired product. Any chemist who has spent some time in a laboratory attempting to synthesize molecules knows that chemical reactions often behave in unwanted ways:
- An undesired product is obtaind instead of the desired one;
- The product yield is too low for the reaction to be useful;
- No reaction happens at all.
In many cases, several synthesis attempts (sometimes dozens or hundreds) are necessary until the desired molecule is eventually obtained, which is a tremendous waste of time and resources.
Coley et al. have published an article titled "A graph-convolutional neural network model for the prediction of chemical reactivity" (Chem. Sci. 2018, 10, 370-377), in which they present an algorithm that is capable of predicting the major product of a chemical reaction correctly in over 85% of the cases that were tested. Such an algorithm can be used to validate synthesis ideas before performing them in a laboratory, thereby preventing a large number of synthesis failures.
The developed method is based on a graph representation of molecules: atoms constitute the nodes of the graph, while bonds constitute the edges of the graph. Predicting the outcome of a reaction is then equivalent to predicting graph edits, ie. which graph edges (representing chemical bonds) are changed.
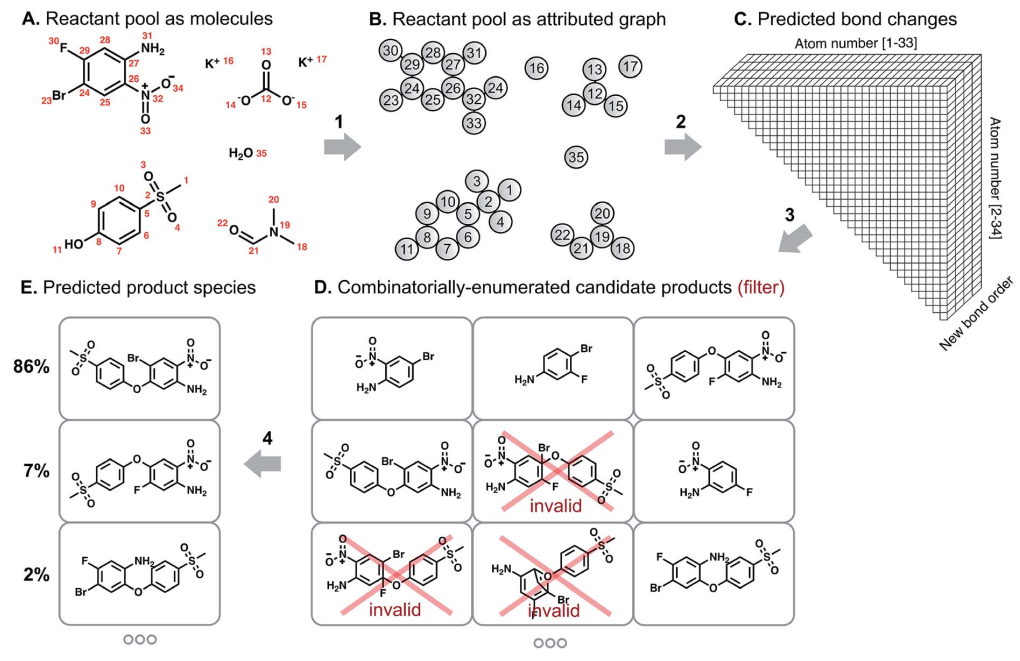
The algorithm developed by Coley et al. mimicks how chemist predict reaction outcomes, as presented in the figure below. First, a graph-convolutional network tries to identify the reactive sites, ie. atoms that are most likely to undergo a change in connectivity (arrow 2). Then, the products that could result from these change in chemical bonding are enumerated (arrow 3), and filtered to eliminate molecules that don't satisfy chemical valence rules (eg. carbon atoms with 5 bonds). Finally, another graph-convolution neural network scores the filtered molecules to yield a probability distribution over these possible reaction products (arrow 4).
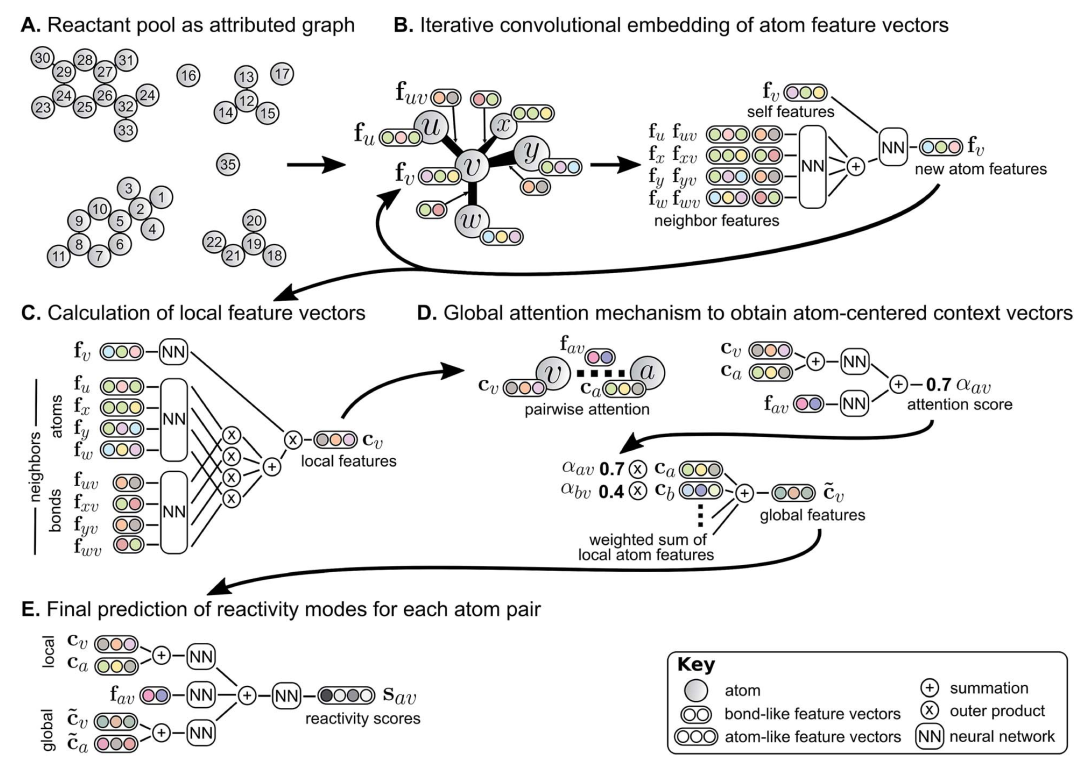
Let's dig deeper into the details of the algorithm. The authors used a Weisfeiler-Lehman Network, a type of graph-convolutional neural network, as depicted in the figure below. First, for each atom-atom pair (including pairs of atoms that are not bound or are located in different reactants) the neural network predicts the likelihood of the bond order to change. The starting point is from a graph representation of reactants (A), where atoms are featurized by atomic number, formal charge, degree of connectivity, valence and aromaticity, and bonds are featurized by bond order and ring status. These atom-level features are iteratively updated (B) by incorporating information from neighbor atoms. A global attention mechanism ensures that information about possible interatctions with atoms that are distant, ie. located in other reactants, are taken into account. This is especially important in order to take into consideration the effect of activating reagents, eg. acids or bases. The next step (C) involves the combination of the atomic features obtained in the step B and bond features to obtain local feature vectors. Then, in step D, the global attention mechanism produces atom-centered context vectors that take into account the effect of distant atoms (eg. in reagents). The local and global context vectors are finally combined (E) to obtain a probability of bond order change for each atom of the system.
The probabilities of bond order changes are used to enumerate candidate products. The authors restricted this enumeration to products that can be obtained by changing up to 5 unique bonds, and taking into account only the most likely bond changes. After this enumeration step, the obtained molecular structures were filtered using chemical valence rules to only keep molecules that can physically exist.
The last part of the algorithm is the ranking of the candidate products. For this task, a second graph-convolutional neural network was used. Candidate outcomes produced by combination of more likely bond changes are themselves more likely to be the true outcome. Quantitative scores for each bond change (from the first part of the algorithm) provides an initial ranking of candidate reaction outcome, which are then refined by taking into account the likelihood of each set of bond changes.
In the end, a probability distribution is obtained. The candidate with the highest probability is likely to be the main product of the reaction. Interestingly, candidates with a lower predicted probability can sometimes help identify impurities that are produced by secondary reactions.
To train the algorithm, the authors used a publicly available dataset, that was constituted from United States patents and comprises almost 410 000 reactions.
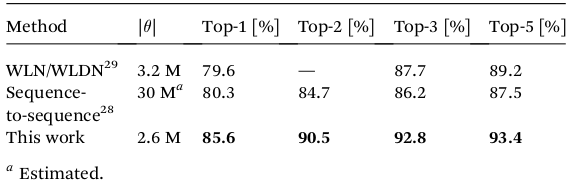
The table below compares the prediction performance of the algorithm with previously-developed methods. For the algorithm presented in this article, the candidate with highest probability (Top-1) was the true reaction product in 85.6% of the cases, which is 5% higher than the previous best algorithm ("Sequence-to-sequence"). In addition to that, the true reaction product was in the 5 candidates with the highest probabilities 93.4% of the time.
A small-scale human benchmarking experiment was performed with 80 questions that were asked to a dozen of chemists of different expertise levels. Globally, the algorithm performed as well as expert chemists.
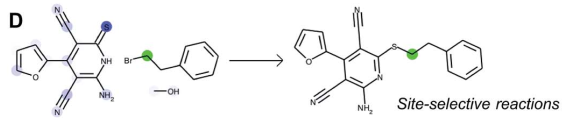
Let's now look at some examples of predictions that were made by the algorithm. In the example D below, the algorithm predicts the correct regioselectivity. The attention mechanism can be examined as follows: one atom which is affected by the reaction is selected and highlighted in green (here, the carbon atom adjacent to the bromine atome). The attention is depicted in different shades of blue: the darker the blue, the higher the attention, meaning a strong influence of this atom on the atom highlighted in green during the reaction. Here we can see that the sulfur atom clearly has the highest attention relative to the carbon atom highlighted in green.
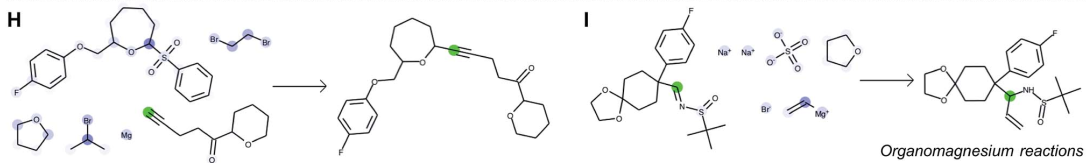
In the examples H and I, the algorithm distinguished between two different uses of an organomagnesium reagent. In the reaction H, the organomagnesium reagent is used as a base to deprotonate the alkyne. In contrast, in the reaction I, the organomagnesium is used as a nucleophile that adds onto the imine substrate.
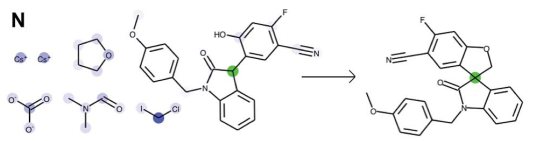
In the example N, a complex formation of a quaternary carbon center is correctly predicted.
By combining two graph-convolutional networks, one to predict the reactive sites and another to rank the candidate products, authors developed an algorithm that is able to reach a predictive power comparable to expert chemists. Interestingly, the two distinct steps of the algorithm and the attention mechanism allow us to gain information about the way the algorithm makes its predictions for a given reaction.
One last remark: it should be noted that this algorithm does not take into account process parameters such as temperature, that can significantly change the outcome of a reaction.