Deep-learning-based inverse design model for intelligent discovery of organic molecules
Discovering new organic molecules that possess a given property is not an easy task. It is a process that is iterative, experiment-intensive and tedious. Furthermore, designing large numbers (let's say hundreds or thousands of them) of new molecules for an application is extremely challenging, even for the most creative chemists. In our last blog post, we presented an AI-based tool that is able to automatically design new molecules, based on a continuous representation of molecules.
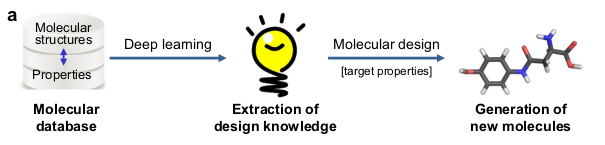
Today, we discuss an article titled "Deep-learning-based inverse design model for intelligent discovery of organic molecules" (npj Computational Materials 2018 4:67), written by Kim et al., that presents a different strategy for discovering molecules, that was developed in a research project by Samsung researchers. The goal of these researchers was to discover new host materials for blue phosphorescent organic light-emitting diodes.
The concept of the study, presented in the following scheme, starts with the assembly of a database that contains thousands of molecular structures and their associated properties. This database is then used by a deep learning algorithm that extracts design knowledge from it (relations between the structure of a molecule and its properties). Using this design knowledge, the algorithm then generates new molecules that are predicted to have some desired target property.
More specifically, the authors of the article wanted to design molecules possessing triplet state energies (T1) higher than 3.0 eV. They started by assembling a database of around 6000 symmetric molecules, by automatically combining selected molecular fragments (aromatic fragments that are often found in organic light-emitting diode hosts). DFT calculations were performed on these 6000 molecules to obtain their (simulated) triplet state energies. This database of molecular structures and associated T1 energies was then used by the deep learning program that they developed.
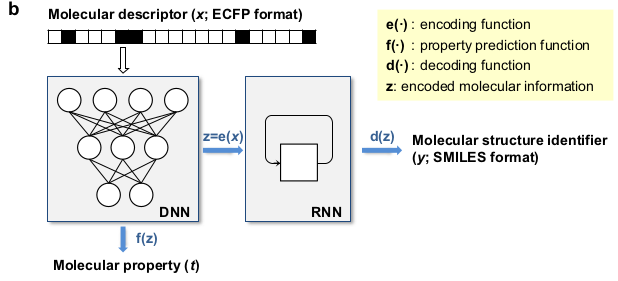
This deep learning program is constituted of three parts, depicted in the following scheme. The first part, in the top left part of the scheme, is a random molecular fingerprint generator (a molecular fingerprint is a binary vector which represents the presence or absence of chemical substructures in a molecule). The second part, the Deep Neural Network (DNN), predicts a molecular property, such as a triplet state energy T1, from a molecular fingerprint. This Deep Neural Network has been trained to predict triplet state energies T1 from molecular structures (that can be easily converted to molecular fingerprints) by using the database. The third part of the program, the Recurrent Neural Network (RNN), is able to decode the output of the last hidden layer of neurons of the DNN into a SMILES string, which corresponds to a molecular structure.
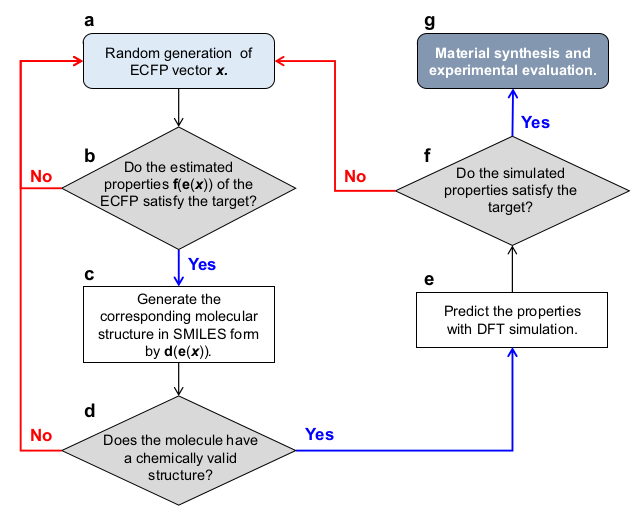
The program functions the following way:
- It generates a random molecular fingerprint;
- The DNN predicts if the molecule corresponding to this fingerprint satisfies the property target (triplet state energy T1 higher than 3.0 eV);
- If yes, the RNN generates the SMILES string of the molecule;
- The program checks if the generated SMILES string is valid;
- If yes, DFT calculations are performed to verify the prediction of the DNN;
- If the DFT calculations confirm the prediction of the DNN, then the molecule can be synthesized and evaluated experimentally.
From 40000 randomly generated fingerprints that were predicted to have triplet state energy T1 higher than 3.0 eV by the DNN, the RNN decoded around 36600 valid SMILES strings. From these valid SMILES strings, 3205 new unique molecules (obtained by removing duplicates and SMILES that were already present in the training data) were obtained.
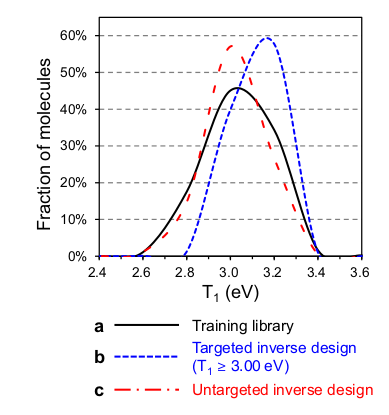
On the following figure, we see that the target of T1 >= 3.0 eV is not reached for all the generated molecules (blue curve), but that the proportion of molecules with T1 >= 3.0 eV is significantly higher than in the training library (59% vs. 36%). Hence, this strategy has successfully oriented the algorithm towards a desired property.
Let's look at some examples of generated molecules in the figure below. For each molecule the T1 energy predicted by the Deep Neural Network and the T1 simulated by DFT are shown. Three categories of molecules are shown:
- Asymmetric molecules constituted of fragments that were already present in the training library, but that are now assembled in a different way;
- Symmetric molecules containing at least one fragment which was not present in the training library;
- Asymmetric molecules containing at least one fragment which was not present in the training library.
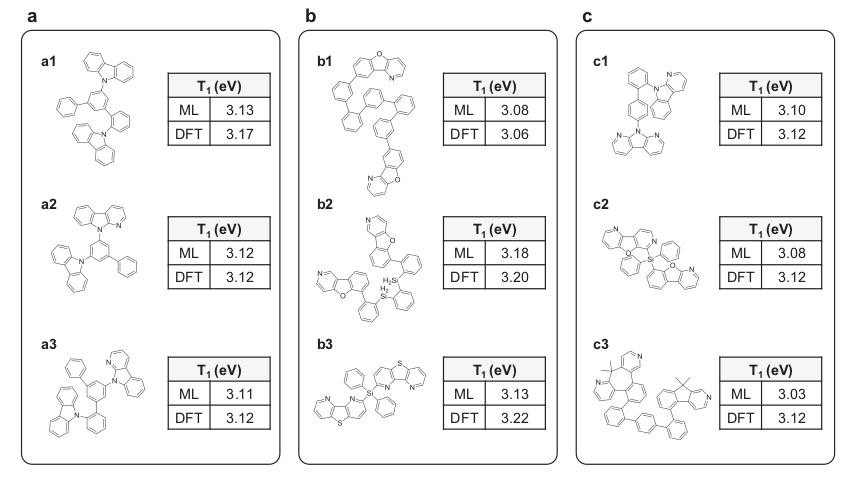
Three molecules (one of each of these categories) were then synthesized and evaluated experimentally: a1, b1 and c1, shown in the figure above.
To sum up, Samsung researchers developed a new way of automatically designing molecules with targeted properties and applied this methodology to the discovery of new molecules for organic electronics.
This method is significantly different from conventional approaches, where molecules are designed first (using rules, heuristics and human knowledge) and the properties of these molecules are predicted using artificial intelligence to only keep the molecules that possess the right properties. Here, the method is fully data-driven, which ensures that humans do not restrict the algorithm's creativity by enforcing design rules.
There is a significant effort to make at the beginning of such a machine learning project, when the initial dataset must be assembled. Here, it involved generating thousands of molecules by automatically combining molecular fragments, and then performing DFT calculations on each of these molecules. However, once this substantial initial effort has been made, the return on investment is high. Additionally, the 3205 molecules that have been automatically designed during this project can be added to the database, which will thus grow and take value over time.