Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules
Designing molecules that have desired properties is a long and difficult process. To obtain a new molecule that can be used in some application, scientists must use their creativity and domain knowledge to propose many new molecules, synthesize them and test them for the given application. Moreover, human creativity often has its limits in the number and diversity of ideas that it can generate.
In the article "Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules" (ACS Cent Sci. 2018, 4, 268-276, Gómez-Bombarelli et al. used machine learning algorithms to automatically design molecules and simultaneously predict and optimize some of their properties.
They used an autoencoder, as illustrated in the diagram hereafter, which is constituted of an encoder and a decoder. The encoder converts a molecule (represented as a SMILES string) into a continuous probabilistic representation. This continuous representation of molecules constitutes the latent space, which is of lower dimensionality than the starting space (thus, the input molecule is compressed into its latent representation). The decoder is able to generate a molecular structure (as a SMILES string) from any point in the continuous latent space. Additionally, the latent representation of a molcule can be used to predict its properties, such as its drug-likeness or synthetic accessibility, using a neural network.
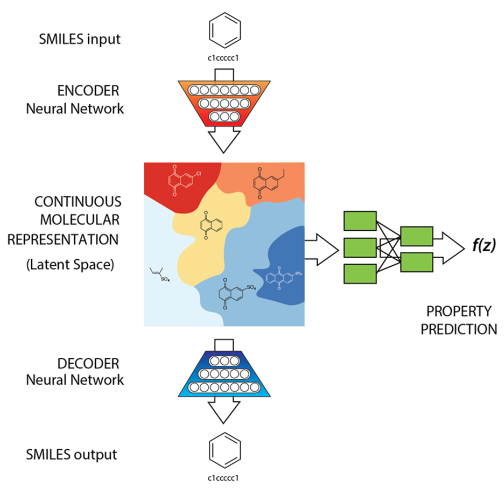
The main interest of compressing molecules to a continuous latent representation is that points that are close to each other in the latent space represent molecules that are similar chemically. This allows to do several interesting things that we will discuss hereafter.
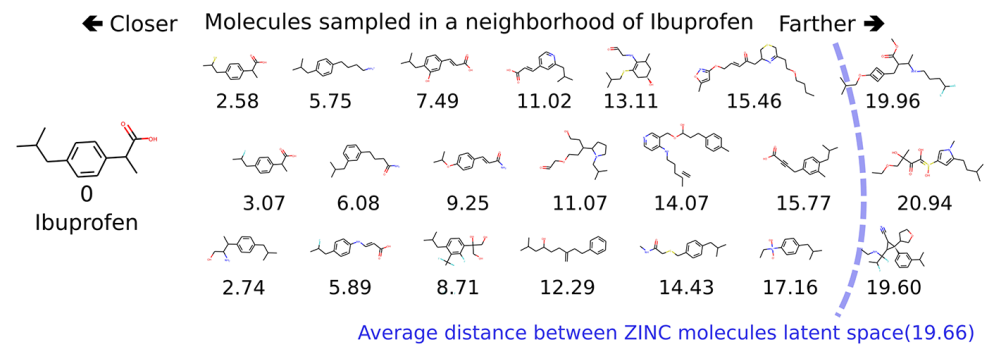
If we convert one molecule to its latent representation and then sample points that are nearby in the latent space, we obtain molecules that are analogs of the starting molecule. For example, in the next figure, this methodology is applied to Ibuprofen (drawn on the left). The other molecules are sampled from the neighborhood of Ibuprofen in the latent space. Under each molecule is written its Euclidian distance from Ibuprofen in the latent space. The molecules on the left are closer to Ibuprofen (both chemically and in the latent space) than molecules further on the right.
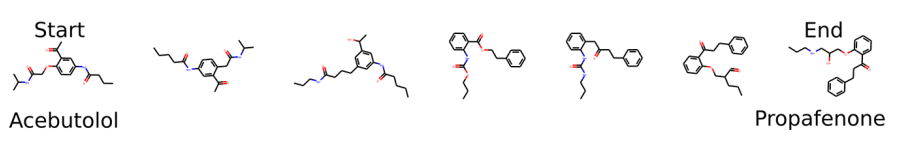
We can also convert two molecules to their latent representations, which gives us two points in the latent space, and then interpolate between these two points. The following figure is an example of interpolation of six molecules between acebutolol and propafenone. The more on the left a molecule is, the more similar to acebutolol it is, and the more on the right, the more similar to propafenone it is.
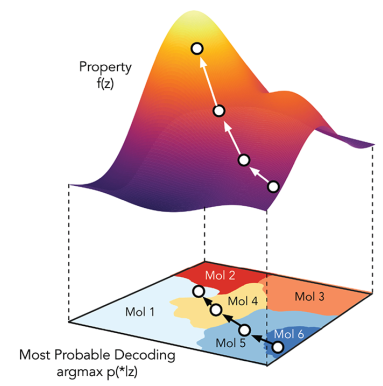
Finally, by training a neural network to predict properties of molecules from their latent representation, it is possible to optimize the structure of molecules to obtain certain properties, as depicted in the following figure. In this figure, we start from molecule 6 and want to maximize its property f(z). The algorithm uses a gradient-based optimization algorithm to progressively move in the latent space to points where f(z) is higher, by following the gradient of f(z). The algorithm finally obtains molecule 1, which has a higher f(z) than the starting molecule 6.
The continuous representation of the molecules is the key feature of this methodology, which allows to easily navigate in the latent space. The combination of the autoencoder algorithm with the predictive neural network is a strength of this article, because it allows to orient the search in the latent space. Indeed, we are generally interested in designing molecules with certain molecular properties, and not just designing molecules without any goal.
In summary, the authors used a continuous representation of molecules to explore the chemical space and automatically generate new molecules. They coupled the autoencoder with a neural network whose task was to predict properties of the molecules represented by points in the latent space. This predictive neural network was used to perform gradient-based optimization of molecular properties. The developed tool will be a valuable complement to the creativity of chemists and allow them to explore the chemical space further and faster.