Using Machine Learning To Predict Suitable Conditions for Organic Reactions
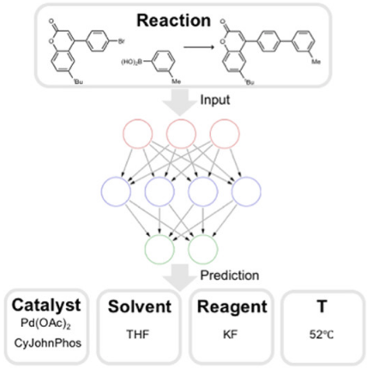
Finding the right experimental conditions (solvent, temperature, catalyst, additives, etc.) for a reaction can be a very time-consuming process. Gao et al. have published an article titled "Using Machine Learning To Predict Suitable Conditions for Organic Reactions" (ACS Cent. Sci. 2018, 4, 1465-1476), in which they present a neural network model that is able to predict appropriate reaction conditions for any organic reaction, including a catalyst, solvents, reagents and the temperature.
In their introduction, the authors note that state-of-the-art methods for predicting reaction conditions for organic reactions are limited because:
- They are often not applicable to large sets of reactions with a sufficient accuracy (eg. because they were developed for specific classes of reactions),
- They do not take into account the compatibility and interdependence of temperature and chemical context (catalyst, solvents, reagents) at the same time,
- They have not been quantitatively tested on large reaction datasets.
The authors designed a hierarchical neural network and trained it on 10 million reactions extracted from the Reaxys database to predict the catalyst, solvents (up to two), reagents (up to two) and temperature for any organic reaction. The reactants and products were represented using their Morgan circular fingerprints, while catalysts, solvents and reagents were represented as one-hot vectors. An important detail is that because of the hierarchical structure of the neural network model, the predictions of the different conditions are made sequentially so that they are compatible: the catalyst is predicted first, followed with both solvents, followed with both reagents, followed by the temperature. That way, the predicted solvent is compatible with the catalyst and the temperature is compatible with all chemical species, for example.
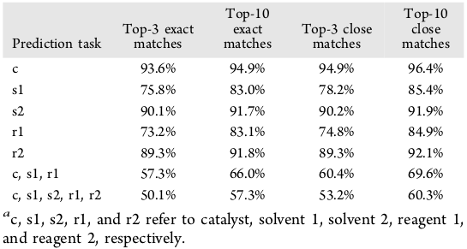
After training, the model was tested on 1 million reactions that were not present in the training set. In the following table are reported the prediction accuracies for the catalyst, the solvents, the reagents, and combinations of these parameters. In the first column are reported the Top-3 exact match frequencies, which are the percentages of reactions where the recorded catalyst (or solvent, etc.) was in the top-3 predicted catalysts. We see that the catalysts are accurately predicted more than 90% of the time. The first solvent and first reagent are less accurately predicted (around 75%). The second solvent and second reagent (or absence thereof) are predicted accurately around 90% of the time, which can be explained by the fact that in many reactions there is no second solvent or reagent. Finally, the full reactions conditions (catalyst, two solvents and two reagents) are accurately predicted around 50% of the time in the top-3, compared with 4.7% in a null model.
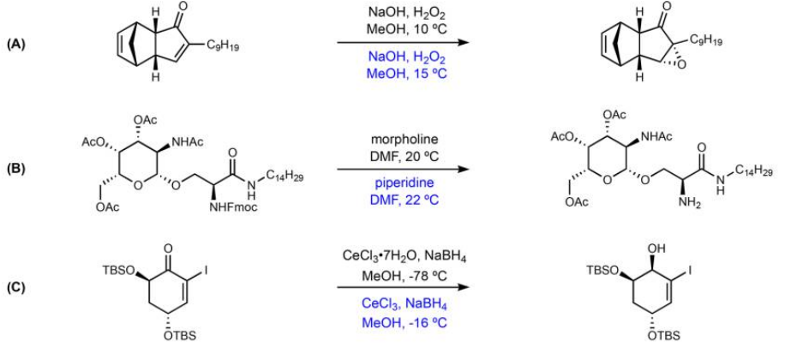
In the figure below, some examples of successful predictions are represented. The reaction conditions of the original article are written in black and the predicted reaction conditions are written in blue. In the example A, an epoxidation reaction, the conditions are correctly predicted. In the example B, a Fmoc deprotection, the predicted reagent, piperidine, is different from the recorded reagent, morpholine, but is chemically very similar. In the example C, a Luche reduction, the neural network correctly recognizes the necessity of using cerium chloride to selectively reduce the carbonyl group.
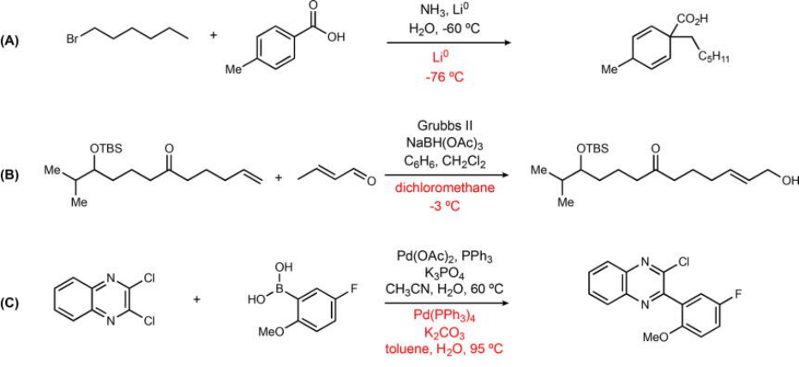
In the next figure are presented reactions where the model gave an incorrect prediction. In the example A, a Birch alkylation, the model does not predict the need to use ammonia. In the example B, the model fails to recognize that this is a two-step reaction (alkene metathesis followed by aldehyde reduction), and thus fails to predict the catalyst and reagent in the top prediction. This issue is probably caused by the representation of this two-step reaction as a one-step reaction in the database used for training. In the example C, the prediction is very reasonable, since a Pd(0) complex is predicted as catalyst, whereas in the recorded reaction a Pd(II) salt was used with in situ reduction. In many examples of incorrect results, the predicted reaction conditions are still quite chemically reasonable for the reaction.
The following two figures are embeddings of the most common 50 solvents and reagents projected on two-dimensional spaces using the t-distributed Stochastic Neighbor Embedding (t-SNE) algorithm, a tool used to visualize high-dimensional data. These t-SNE embeddings were constructed from the weight matrix in the last hidden layer of the neural network. In the case of solvents, two solvents that have similar weigths in the neural network will have similar scores in the reaction conditions predictions and will be located close to each other in the 2-dimensional embedding. In the figure below, we can clearly see that chemically similar solvents are clustered together.
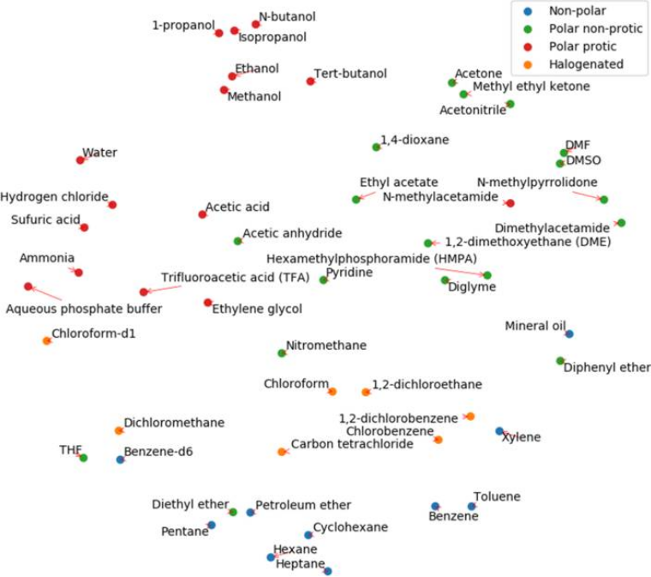
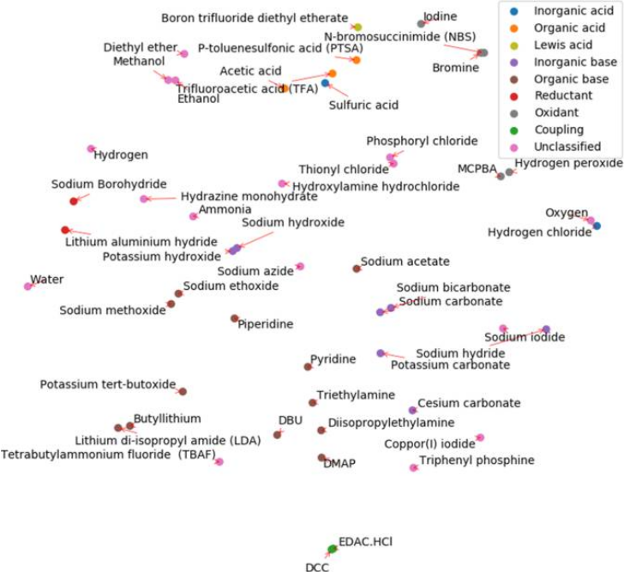
In the reagents embedding below, chemically similar reagents are also clustered together, meaning that their associated weigths in the neural network are similar.
To sum up, the authors of this study have developed a neural network algorithm that is capable of predicting reaction conditions for any organic reaction. They have trained it on a large dataset (10 million reactions) and tested it on 1 million reactions. The algorithm is able to recover the catalyst, at least one solvent and on reagent that are close to the reported ones (in the literature) in the top-10 predictions for 70% of the tested reactions. This score can certainly be improved, but the article sets a serious benchmark for future developments in this field.
Since it has been trained on 10 million reactions, this new model has a better capacity for generalization than previous models. Let's note that concentration is not included in the model because it is rarely included in the database. A point of caution: the algorithm does not take into account safety hazards! Thus, the predicted reaction conditions should always be validated by a chemist before being run in the laboratory, to exclude hazardous reaction conditions.