Machine learning descriptors for molecules
Using machine learning to predict properties of molecules or to design molecules with desired properties plays an important role in the field of chemistry, toxicology and materials science. In machine learning, when it comes to predicting the properties of molecules, it is necessary to convert the molecular structures into values that can be used by machine learning algorithms.
The molecular structures are transformed into descriptors before being used to train the machine learning model, as depicted in the illustration below. Usually, a descriptor is a number, a vector or a matrix, but other data formats such as character strings are possible.
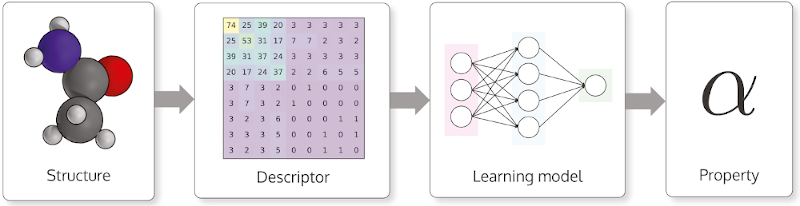
Typical workflow to make machine learning predict a property from a molecule. Source: DScribe: Library of descriptors for machine learning in materials science.
The purpose of this article is to give an overview of descriptors that can be used for molecules and to address three main questions:
- What makes a descriptor appropriate for a machine learning problem?
- What are the different types of descriptors?
- How to select descriptors?
What makes a descriptor appropriate for a machine learning problem?
When selecting a descriptor to encode molecules, it is important to ensure that the three following criteria are met:
The descriptor is correlated with the properties of the molecules that are to be predicted.
The more correlated are the input part of the data with the output part of the data given to a machine learning algorithm, the better the predictive model will be. In the case of molecules, the data used to train the machine learning algorithm are the descriptors and the properties of the molecules. Therefore, a descriptor that has a low correlation with the properties of the molecules to predict will give a model with a poor predictive performance.
The descriptor generates distinct values for structurally different molecules, even if the structural differences are small.
When using a descriptor that does not generate dissimilar values for structurally different molecules, some information about the features of the molecules is lost. This loss of information directly affects the performance of the machine learning algorithm.
The descriptor is adapted to the size of the molecules that are used in the machine learning algorithm.
Not all descriptors are suitable for all sizes of molecules. Some descriptors are only useful when applied to small molecules, whereas other descriptors are defined specifically for large molecules such as polymers and proteins. If a descriptor is not adapted to the size of the molecules, the descriptor may not represent the molecular features accurately or may give a representation of the molecules too complex for the machine learning algorithm. In these cases, information about molecular features is lost. For example, SMILES are suitable descriptors for small molecules, but not for large molecules such as polymers. In fact, polymers are stochastic molecules and, therefore, they do not have unique SMILES representations. In this case, a suitable descriptor can be the “BigSMILES”. Presented by Tzyy-Shyang Lin et al. in the article "BigSMILES: A Structurally-Based Line Notation for Describing Macromolecules", this descriptor aims to provide representations that can be used for polymers.
Depending on the machine learning problem, other important criteria can be required for the descriptor. For example, a descriptor may need to:
- Differentiate isomers. For example, cis/trans isomers are handled by using geometrical descriptors.
- Be decodable (back from the descriptor to the molecular structure). This characteristic is essential if the machine learning project aims to generate molecules. In this case, SMILES are a possible descriptor. An example of the application of SMILES to chemical design can be found in the article "Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules". In this article (which we have presented in a previous blog post), Gómez-Bombarelli et al. used SMILES in a machine learning project to automatically design molecules and simultaneously predict and optimize some of their properties.
In addition, two other points should be considered when selecting descriptors: the amount of data and the dimension of the descriptor(s).
The amount of data required in a machine learning project is highly related to the complexity of the problem. For a small dataset, high-dimensional descriptors are not recommended. In fact, high-dimensional descriptors increase the dimensionality of the problem and thus make the data sparser. As a result, training the machine learning model becomes more complex and therefore more data are needed to obtain a model with a satisfying predictive performance. In general, the number of molecules in the dataset should be much higher than the number of dimensions of the descriptors.
What are the different types of descriptors?
Molecular descriptors can be classified in two categories: the experimental descriptors and the theoretical descriptors.
Experimental descriptors are any physicochemical properties obtained by experimental measurements or by numerical simulations. For example, the partition coefficient P is commonly used as a molecular descriptor in medicinal chemistry and drug discovery. The partition coefficient is the ratio of the concentrations of a compound in a mixture of two non-miscible solvents at equilibrium.
Theoretical descriptors are derived from the symbolic representation of the molecules, such as the structural formula or the empirical formula. This category of descriptors can be divided in five classes. The following illustration represents the five classes of theoretical descriptors and the relationship between their dimensionality, the information they provide and the ease of calculation.
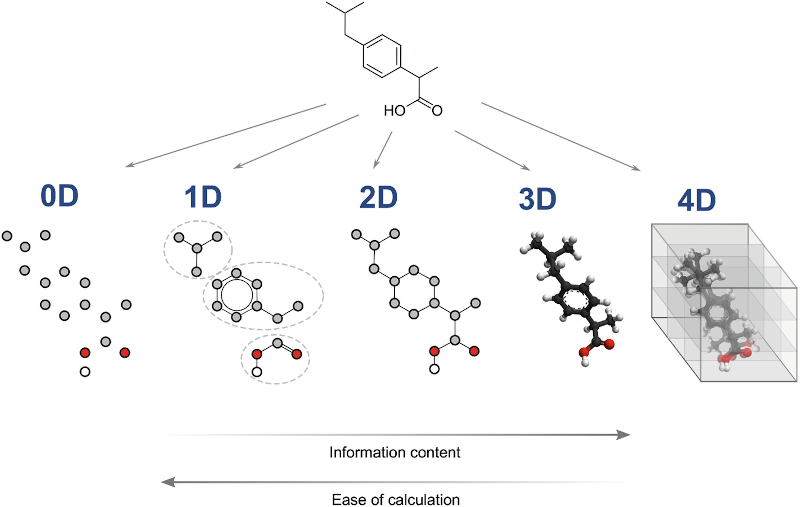
Representation of the five classes of theoretical descriptors and the relationship between their dimensionality, the information they provide and the ease of calculation. Source : Molecular Descriptors for Structure–Activity Applications: A Hands-On Approach.
- 0D descriptors. This class regroups all molecular descriptors that do not provide any information about the molecular structure or connectivity of atoms. For example, atom counts, bound counts or molecular weights are 0D descriptors. An advantage of these descriptors is that they are easily obtained. However, took one by one, these descriptors do not contain much information about the structural features of the molecules and, therefore, they are often combined with other descriptors.
- 1D descriptors. This class regroups the molecular descriptors that can be calculated from a set of substructures such as functional groups. The most common 1D descriptors are the fingerprints. For example, a fingerprint can be a binary vector where 1 indicates the presence of a structural feature and 0 its absence. Like 0D descriptors, 1D descriptors are easily obtained.
- 2D descriptors. This class regroups all descriptors that provide information on molecular topology based on the graph representation of the molecules. Typical 2D descriptors are the adjacency matrix, the Coulomb matrix or the distance matrix. In the case of the adjacency matrix, the descriptor indicates which atoms are bonded in a molecule. Because the 2D descriptors are sensitive to structural features of the molecule (size, shape and symmetry), they are a common choice as molecular descriptors.
- 3D descriptors. This class regroups all geometrical descriptors that provide information about the spatial coordinates of atoms of a molecule. The most well-known 3D descriptors are the molecular matrix and the 3D-MoRSE descriptors. In the case of the molecular matrix, the descriptor represents the Cartesian coordinates (x, y, z) of each atom. These descriptors provide a lot of information about molecules and have the advantage of differentiating isomeric molecules, which is not the case for all descriptors. However, because of their complexity, the geometrical descriptors can be time-consuming to calculate.
- 4D descriptors. The 4D descriptors are also called "grid-based descriptors". These descriptors, in addition to the molecular geometry, introduce a fourth dimension. This new dimension usually characterizes the interactions between the molecule(s) and the active site(s) of a receptor or the multiple conformational states of the molecule(s). Common 4D descriptors are CoMFA and GRID. An advantage of the 4D descriptors is that they provide more information than the other descriptors and are always able to generate dissimilar values for structurally different molecules. However, like the 3D descriptors, the 4D descriptors are not easy to obtain because of their higher complexity.
How to select descriptors?
With thousands of existing molecular descriptors, selecting the most suitable descriptors is a difficult task.
When several descriptors are suitable for a machine learning problem, there are two main strategies for choosing between the descriptors: the exhaustive search and the optimization algorithms.
The exhaustive search, also known as the All Subset Model (ASM), is the simplest strategy. For a set of descriptors, it consists of the generation of all possible combinations of descriptors (from a single descriptor to a combination of descriptors). The ASM guarantees that the best set of descriptors is found but it can be very computationally consuming depending on the number of descriptors to test. For example, for N descriptors to test, 2^N-1 combinations of descriptors are tested. This strategy is recommended if there are few descriptors to test.
Optimization algorithms are iterative procedures that can be used to find the optimal combination of descriptors from a set of molecular descriptors (that leads to the best predictive model for the studied properties). The most common optimization algorithms used in the selection of the descriptors are evolutionary programming (EP), ant colony optimization (ACO), sequential search (SS) and genetic algorithms (GAs). The Illustration 3 shows the general workflow of the optimization of the descriptors selection.
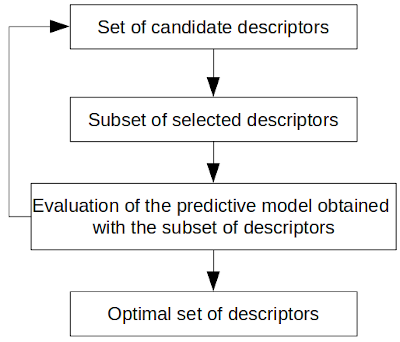
Worflow of an optimization to select the optimal subset of descriptors.
Starting from a set of descriptors to test, the optimization selects a subset of descriptors and evaluates the performance of the predictive model obtained with it. This step is repeated until an optimal set of descriptors is found or until a stopping criterion is reached.
An application of GAs for molecular descriptors selection can be found in the article "QSAR study of the calcium channel antagonist activity of some recently synthesized dihydropyridine derivatives. An application of genetic algorithm for variable selection in MLR and PLS methods". In this article, Hemmateenejad et al. performed a QSAR (Quantitative structure activity relationship) study on 72 diester-substituted dihydropyridine derivatives by using MLR (Multiple Linear Regression) and PLS (Partial least squares) procedures. Starting from more than 240 theoretical descriptors, the best set of descriptors was successfully selected by the GA method and was reduced to an optimal set of 18 descriptors.
Conclusion
To be usable in a machine learning project, molecules must be represented by descriptors. The choice of a descriptor is important because it has a strong influence on the predictive performance of a model.
There are many descriptors and some of them have specificities (for example, the ability to be differentiate isomers) but when selecting descriptors, the four main concerns are the correlation between a descriptor and the properties to predict, the capacity of a descriptor to generate dissimilar value for structurally different molecules, the number of dimensions of the descriptors and the amount of data available.
Descriptors cannot fully describe the structural complexity of a molecule. This issue can be partially solved by using high-dimensional descriptors or multiple descriptors. Nevertheless, it should be kept in mind that using high-dimensional descriptors or multiple descriptors increases the complexity of the problem and can, therefore, reduce the performance of the machine learning algorithm.