Optimizing Chemical Reactions with Deep Reinforcement Learning
Optimizing chemical reactions is a very common task for chemists. It usually aims at maximizing the yield or selectivity of a reaction in order to get the most possible product from some raw material.
The traditional approaches used by chemists to optimize reactions involve changing one reaction parameter at a time (eg. temperature) while keeping the other parameters constant (eg. concentrations, pressure and reaction time), or search exhaustively all combinations of reaction conditions (which is obviously very time-consuming). While these approaches work pretty well, they can be quite slow to reach optimal reaction conditions.
Alternative reaction optimization methods have been designed, such as the Stable Noisy Optimization by Branch and Fit (SNOBFIT) method. They are usually more rapid than optimizing one variable at a time, but there is still room for improvement.
In the article "Optimizing Chemical Reactions with Deep Reinforcement Learning" (ACS Cent. Sci. 2017, 3, 1337−1344), Zhou et al. used Deep Reinforcement Learning to automatically optimize chemical reactions.
Reinforcement learning is a class of machine learning algorithms, where the algorithm tries to reach some goal (in our case, maximizing the reaction yield) by performing actions step-by-step (in our case, performing chemical reactions with certain reaction parameters), and recieves a reward each time it gets closer to its goal. The closer to the goal the algorithm gets at one step, the larger reward it recieves. This reward system allows the algorithm to learn what are the more pertinent actions to perform in order to optimize chemical reactions and to get better over time.
The algorithm presented in this paper, called Deep Reaction Optimizer, is based on a kind of Recurrent Neural Network (RNN) called Long Short-Term Memory (LSTM). It is capable of remembering a sequence of past actions to predict the next best action. In the Deep Reaction Optimizer, the algorithm uses the sequence of all the reactions that have been performed to suggest a next reaction with a higher predicted yield. It tries to balance between exploitation (try to reach a higher yield by suggesting reaction conditions close to the current conditions) and exploration (suggest reaction conditions that are significantly different from the current conditions) in order not to get stuck in a local yield maximum.

Machine learning algorithms always require to be trained before being used. This can be a problem when we don't have existing data to train our algorithm. The authors circumvented this problem by pre-training their algorithm on simulated non-convex mathematical functions that possess several local maxima and one global maximum, and thus mimick response surfaces of chemical reactions.
After this pre-training step, the authors used their algorithm to optimize organic reactions performed in an electrospray. The parameters that were varied for optimization were :
- The flow rate of the reactants solution in the electrospray;
- The voltage;
- Pressure.
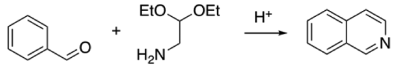
Four organic reactions were optimized. The first one, the Pomerzanz-Fritsch synthesis of isoquinoline, is shown hereafter.
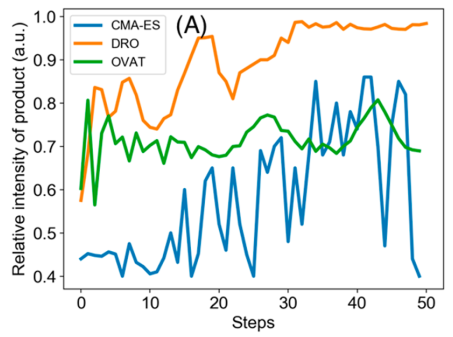
Three optimization algorithm are compared: the Deep Reaction Optimizer (DRO), and two other state-of-the-art algorithms. The DRO is clearly more rapid than the two others to reach a yield maximum, and this maximum is higher. However, given that the reaction is performed in an electrospray, it is not possible to know the yield that has been reached.
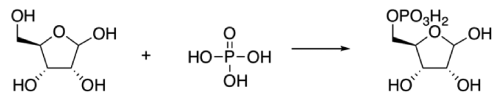
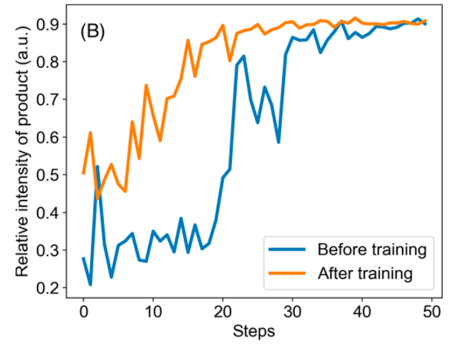
What is even more impressive is that this algorithm gets better at each reaction it optimizes! When optimizing the synthesis of ribose phosphate (shown hereafter), an algorithm that has already been used to optimize two other reactions (including the Pomeranz-Fritsch Synthesis of Isoquinoline shown above) is faster to reach optimal reaction parameters than an algorithm that has never optimized other chemical reactions. This means that the algorithm learns general principles of chemical reaction optimization, even when the respective reaction mechanisms are very different!
The method developed by the authors is not limited to organic reactions. Indeed, they have applied it to the optimization of the synthesis of silver nanoparticles from silver nitrate. Their objective was to maximize the absorbance at 500 nm, in order to obtain Ag nanoparticles with an average diameter of 100 nm. The optimization parameters were:
- The concentration of sodium borohydride;
- The concentration of trisodium citrate;
- The reaction temperature.
The deep reinforcement learning algorithm successfully found reaction conditions that maximized absorbance at 500 nm. However, what is lacking from this research paper is a characterization of the obtained nanoparticles with a UV-visible spectrum (to confirm that the maximum absorption wavelength is effectively 500 nm) and Transmission Electron Microscopy (to determine the obtained average nanoparticle size and confirm it is around 100 nm).
Another limit of this article is the absence of optimization of organic reactions in classical batch experiments (the organic reactions were only optimized in an electrospray, and not in bulk reactions). Furthermore, how does the deep reaction optimizer behaves when there are more than 3 parameters to optimize? Answering these questions would give even more value to this already very interesting article.